




臨床試驗中的單盲、雙盲、三盲、破盲是什么意思?
盲法試驗常用的有兩種:單盲(single blinding)和雙盲(double blinding),更嚴格的對照試驗要用到三盲(triple blinding),在對照藥物和試驗藥物劑型或外觀不同時,還要用到雙盲雙模擬技


注冊備案 · 臨床試驗 · 體系建立輔導 · 分類界定 · 申請創新
Recommend case

來源:醫療器械注冊代辦 發布日期:2023-11-28 閱讀量:次
藥物臨床試驗必備文件保存至試驗藥物上市后5年,夠了嗎?答案是不一定,下面我們來詳細闡述一下。
")
根據中國2020版GCP,第八十條:用于申請藥品注冊的臨床試驗,必備文件應當至少保存至試驗藥物被批準上市后5年;未用于申請藥品注冊的臨床試驗,必備文件應當至少保存至臨床試驗終止后5年。
根據以上規定,我們可以知道:
1、用于申請藥品注冊的臨床試驗,必備文件應當至少保存至試驗藥物被批準上市后5年,考慮到是中國GCP的規定,所以這里的批準上市應該是指在中國境內注冊上市。
2、未用于申請藥品注冊的臨床試驗,比如上市后的臨床試驗,必備文件保存至臨床試驗終止后5年也是符合規定的。
3、上述規定中未說明是誰來保存,因GCP主要規定了研究者和申辦者的職責,故研究者和申辦者均應至少保存必備文件至試驗藥物被批準上市后5年。
不知大家是否思考過,上述法規條款中規定的是“至少保存至試驗藥物被批準上市后5年”,什么情況下會超過5年呢?舉個例子,如果中國企業研發的新藥在中國境內上市后,還申請在其他ICH成員國上市,則還應該遵循ICH GCP中對于必備文件的保存規定,因為中國也是ICH成員之一:
研究者的職責:4.9.5 Essential documents should be retained until at least 2 years after the last approval of a marketing application in an ICH region and until there are no pending or contemplated marketing applications in an ICH region or at least 2 years have elapsed since the formal discontinuation of clinical development of the investigational product.
申辦者的職責:5.5.11 The sponsor specific essential documents should be retained until at least 2 years after the last approval of a marketing application in an ICH region and until there are no pending or contemplated marketing applications in an ICH region or at least 2 years have elapsed since the formal discontinuation of clinical development of the investigational product.
從上述ICH GCP的規定來看,不管是研究者,還是申辦者,都應該保存必備文件到藥物在最后一個ICH成員國獲批上市或者不再計劃在其它ICH成員國上市后至少2年,或者決定放棄這一藥物開發后至少2年。ICH有3個創始監管機構成員即美國、日本和歐盟,考慮到歐盟有20多個國家,故要達到上述條款中要求的“至少2年”會是一個相當漫長的過程。所以,中國企業研發的新藥在中國獲批上市后,如果還想在其他ICH國家如美國或者歐盟國家注冊上市,則必備文件的保存時間可能遠遠超過“試驗藥物被批準在中國上市后5年”。
其實,我們從保存必備文件的目的出發,可能就更加容易理解GCP和ICH GCP中的規定了。必備文件是指能夠單獨或者匯集后用于評價臨床試驗的實施過程和試驗數據質量的文件。保存必備文件的目的也就是準備監管當局的檢查,以證明“試驗實施過程和試驗數據質量”符合法律法規的要求并能夠支持藥物的上市注冊申請。所以,從這一根本目的出發,再結合GCP的要求和企業的藥物開發策略,從而決定你的臨床試驗必備文件到底要保存多久。
引申開來,我們在學習其他GCP條款時也需要從本質出發,這樣才能更好的理解GCP的要求,進而用以指導平時的工作實踐。
來源:道一

站點聲明
本網站所提供的信息僅供參考之用,并不代表本網贊同其觀點,也不代表本網對其真實性負責。圖片版權歸原作者所有,如有侵權請聯系我們,我們立刻刪除。如有關于作品內容、版權或其它問題請于作品發表后的30日內與本站聯系,本網將迅速給您回應并做相關處理。
鄭州思途醫療科技有限公司專注于醫療器械產品政策與法規規事務服務,提供產品注冊備案申報代理、臨床試驗、體系建立輔導、分類界定、申請創新辦理服務。

盲法試驗常用的有兩種:單盲(single blinding)和雙盲(double blinding),更嚴格的對照試驗要用到三盲(triple blinding),在對照藥物和試驗藥物劑型或外觀不同時,還要用到雙盲雙模擬技

剛接觸CRO行業的小伙伴,在學習文件法規資料的同時,常看到一些英文類專業名詞不知道是什么意思。下面,一起看看常見的臨床試驗專業術語: CRO行業的常用術語解釋: 1:新藥研發

SSU是Study Start Up的縮寫,從最初的項目準備,到啟動訪視(Site Initiation Visit)之前所有的準備工作,對整個臨床研究項目的啟動非常關鍵。負責這個關鍵階段工作的部門人員,就叫做SS

醫學的進步是以研究為基礎的,這些研究在一定程度上賴于以人作為受試者的試驗。--《赫爾辛基宣言》。Ⅰ期臨床研究目的是確定可用于臨床新藥的安全有效劑量與合理給藥方案。根據
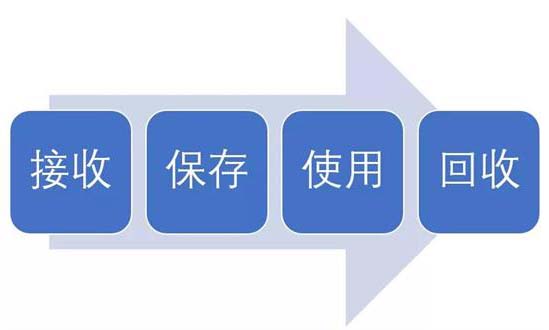
試驗用藥品是指用于臨床試驗的試驗藥物、對照藥品。試驗用藥品滲透到了臨床試驗過程中的每一個步驟,包括藥物的生產、包裝、運輸、保存、使用、回收等。今天我們從臨床試驗中

不少二三類需要臨床的產品,客戶一聽到臨床報價就退縮。既然這么貴,還不如自己做......事實真的是這樣嗎?臨床報價費用都由哪些組成?費用都誰收走了?自己做又有哪些風險?文

隨著醫療器械出口的日益增長,根據市場的需求各醫療器械生產廠商需要符合國家和地區的質量體系法規越來越多,所以經常會碰到出處于不同法規或標準的一些比較容易混淆的概念及

大多數CRO公司在臨床試驗現場啟動會(SIV)上,常由CRA主導。作為一名有上進心的CRA必須清楚的了解到臨床試驗現場啟動考察的流程,再分享一些本人在啟動會考察的細節,請看下文。

俗話說“知己知彼,百戰不殆”,對于作為CRC的我們,自認為對CRA其實已經很了解了,但是在我們工作過程有一個角色平時接觸不到,但是卻又繞不開躲不過,尤其是面對滿屏EDC query的

在臨床試驗中,無論是監查員、質控人員或者項目管理人員到研究中心查看項目資料的時候,總會多多少少發現一些問題,有些問題可能大家都比較熟知,但處理手法五花八門的。處理
行業資訊
知識分享
八年
醫療器械服務經驗
聯系思途,免費獲得專屬《落地解決方案》及報價
咨詢相關問題或咨詢報價,可以直接與我們聯系
思途CRO——醫療器械注冊臨床第三方平臺
北京市 重慶市 河南省 安徽省 天津市 上海市 河北省 陜西省 山西省 內蒙古 遼寧省 吉林省 黑龍江省 江蘇省 浙江省 福建省 江西省 山東省 湖北省 廣東省 廣西 海南省 四川省 貴州省 云南省

微信公眾號
隨時隨地了解行業動態
客服二維碼
詢價咨詢流程 Copyright ? 2016-2026 思途醫療科技有限公司 版權所有丨備案號:豫ICP備2021016482號-1 ![]() 豫公網安備 41010202003160號 網站地圖
豫公網安備 41010202003160號 網站地圖